
科技动态
 打印
打印 催化科学的发展和进步依赖于催化反应机理在原子、分子尺度上的研究和探索。确定反应物与催化剂在真实反应过程中的状态是理解其反应机理的先决条件。近年来,原位技术的进步为表面科学和催化科学不断带来全新的实验现象,激励着理论学家们一方面重新认识经典理论和模型的有效性及其适用范围,另一方面积极思考和探索新的分子机制和理论模型。“实验探秘,理论解密”为人们揭示了许多新奇的原位物理、化学现象。最近,中国科学院上海高等研究院上海光源科学中心高嶷理论课题组助力原位实验研究,在反应环境的动态界面研究方面连续取得了重要进展,其研究成果先后发表在《Science》和《Nature Catalysis》杂志上。
催化科学的发展和进步依赖于催化反应机理在原子、分子尺度上的研究和探索。确定反应物与催化剂在真实反应过程中的状态是理解其反应机理的先决条件。近年来,原位技术的进步为表面科学和催化科学不断带来全新的实验现象,激励着理论学家们一方面重新认识经典理论和模型的有效性及其适用范围,另一方面积极思考和探索新的分子机制和理论模型。“实验探秘,理论解密”为人们揭示了许多新奇的原位物理、化学现象。最近,中国科学院上海高等研究院上海光源科学中心高嶷理论课题组助力原位实验研究,在反应环境的动态界面研究方面连续取得了重要进展,其研究成果先后发表在《Science》和《Nature Catalysis》杂志上。
-
分子尺度观察水分子在二氧化钛表面上的吸附活化和反应
二氧化钛是一种常见的触媒催化剂,可用于光催化和水煤气催化反应。在加热的条件下,可以催化水和一氧化碳产生氢气和二氧化碳。这个反应已经发现近百年了,但是其中分子层面催化剂是怎么催化反应进行的,没有人亲眼见到过。为了实现在原位环境电镜中直接观察到水分子在二氧化钛表面的吸附结构和反应过程,浙江大学和丹麦技术大学的研究人员设计在二氧化钛001型重构表面进行实验。该重构表面有序分布着高活性凸起阵列,吸附在其上的水分子的有序排列为电镜观察提供了充分的衬度。在研究人员的实验中,人们首次观察到吸附水分子呈现一种“双凸起”结构(图1),并在后续的水煤气反应中直接观察到这种“双凸起”结构的动态变化,从而实现了在分子尺度直接观察催化反应进行这一梦想。
为了解析“双凸起”结构的确切原子结构与构成,高嶷研究团队进行了细致充分的第一性原理计算及热力学分析。通过对各种不同二氧化钛表面水结构的构造、计算、和筛选,最终确定了实验观察到的吸附水分子结构:一个水分子在二氧化钛表面打开后形成的两个OH离子和另外两个吸附水分子形成了一种稳定的复合结构(图2)。通过理论计算,研究人员更进一步理解了这种独特的双凸起结构在此前的理论与实验工作中未被发现的原因。原来,这种结构的稳定性依赖于水汽环境的温度与压强,只在接近真实反应条件的水汽环境中才会稳定存在,所以在此前的非原位研究和低压原位研究中都观察不到。在确认了吸附水分子确切结构之后,研究人员最终计算出了在二氧化钛重构表面水煤气反应的完整反应路径,并对实验观察到的双凸起结构动态变化进行了解释。
在这个工作中,研究人员充分展现了理论和原位实验结合得到的催化反应信息的价值,得到了审稿人高度的评价:“This paper does push science forward in terms of the detail of information that can be obtained via very high level electron microscopy coupled with careful chemistry and DFT”。浙江大学袁文涛博士、中国科学院上海高等研究院朱倍恩副研究员、中国科学院上海应用物理研究所李小艳博士生为共同第一作者。中国科学院上海高等研究院高嶷研究员、浙大电镜中心张泽院士、王勇教授、丹麦技术大学的Wagner教授为共同通讯作者。该工作得到了国家自然科学基金委、教育部、科技部、浙江省自然科学基金、上海市自然科学基金、中科院青促会、国家超级计算广州中心、上海超算中心、中国博士后基金的共同资助和支持。
该工作发表在《Science》杂志上,文章链接:https://science.sciencemag.org/content/367/6476/428
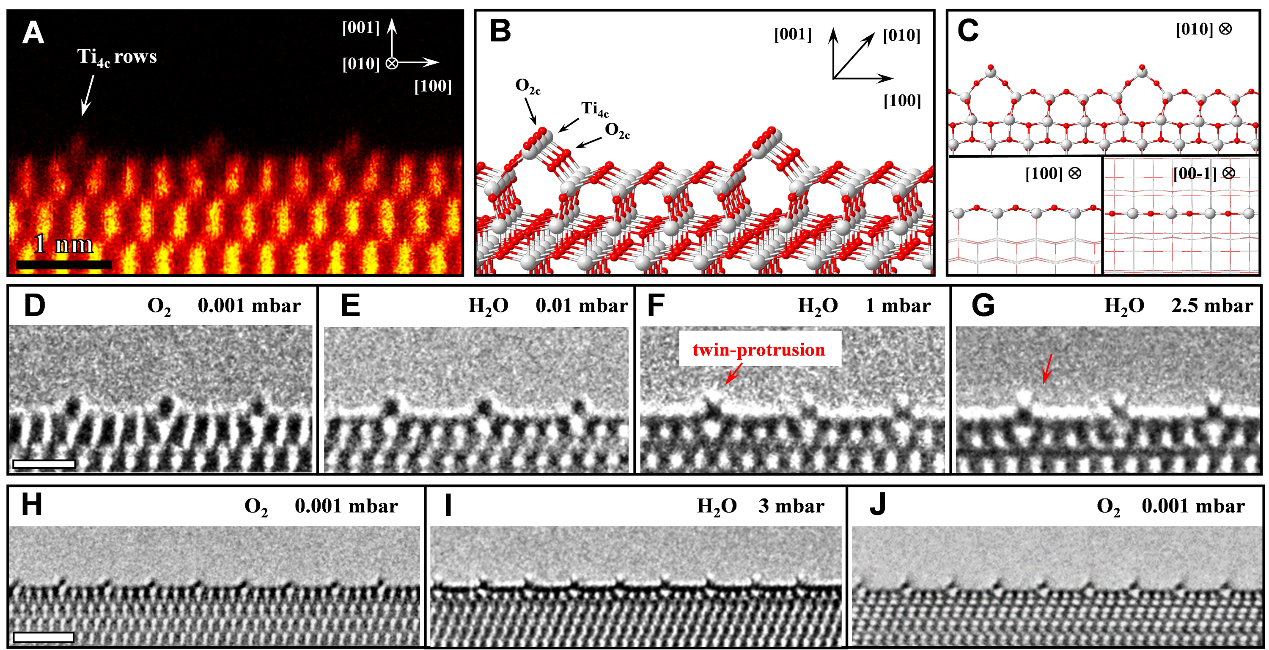
图1,原位实验观察到的二氧化钛表面吸附水分子“双凸起”结构
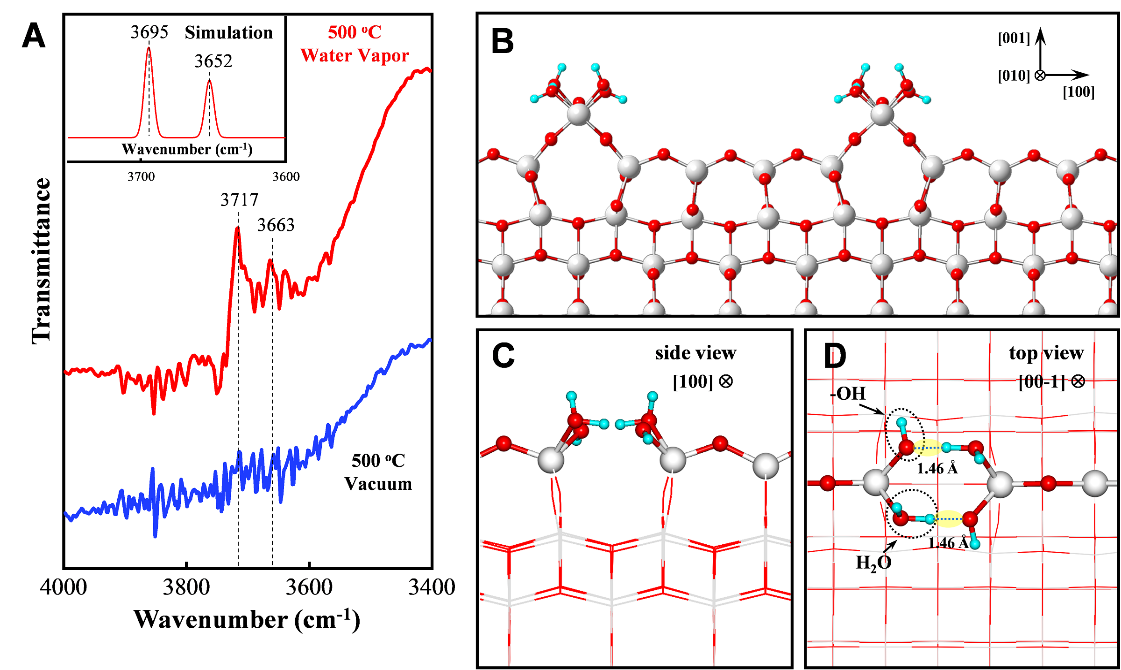
图2,理论计算得到的水分子双凸起原子结构以及理论与实验的红外谱线比较
-
AuNi双金属催化剂核-壳结构在CO2加氢反应中的动态变化
双合金核壳结构催化剂作为一种成功、成熟的可控设计和合成纳米催化剂而广为人知。在过去的研究工作中,人们对核壳结构催化剂的几何以及电子结构及其在催化反应中起到的作用进行了一系列的研究。通常认为,核壳结构催化剂的活性和选择性来源于其独特的核-壳结构。这一认识促发了大量的核壳结构催化剂的合成和研究。然而,最近中科院上海高等研究院高嶷研究员团队与中科院大连化物所刘伟副研究员、杨冰副研究员团队及南方科技大学谷猛副教授团队合作,结合多种原位技术及理论模拟,在原子尺度上解析了镍金双金属纳米催化剂的核-壳结构在反应中的动态演变过程,揭示了该催化剂在二氧化碳加氢反应中的真实表面,并提出了新的催化机理。该工作颠覆了人们对核壳结构催化剂构效关系的固有认识,充分说明了原位研究在催化研究中的必要性和重要性。
该研究团队基于原位透射电镜以及特殊设计的毫巴级负压定量混气系统,研究了镍金双金属纳米催化剂在二氧化碳加氢反应中的结构演变过程。实验中发现甲烷的产物选择性不到5%,高达95%的产物是一氧化碳。利用传统的离线电镜进行观测,发现该催化剂在反应前后都呈现出镍纳米球为内核、2到3个金原子层为壳层的核壳构型。按照常规理解,人们会认为超薄的金壳层包裹了镍导致了高的一氧化碳选择性。然而原位电镜、原位红外和原位X射线吸收谱结果都证明随着反应的进行,催化剂内部的镍原子迁移至表面,导致合金化,壳层逐渐消失;随着反应结束,表面的镍原子又会迁移回催化剂内部,发生退合金化,壳层又会逐渐出现(图3)。
为了理解实验结果,高嶷研究团队对不同反应物以及反应中间产物吸附情况下的金镍合金表面偏析倾向进行了系统研究,结果发现反应中催化剂表面产生的一氧化碳吸附分子是导致镍原子迁移到表面的元凶(图4)。合金化表面上的镍原子提供了二氧化碳加氢反应的活性位点,而表面金原子则提供了一氧化碳脱附位点,导致了95%的反应选择性。反应结束之后,由于表面一氧化碳都可以通过表面金原子脱附,镍原子就逐渐又进入催化剂内部。金壳层会在催化剂表面重新形成。理论计算提出了新的核壳结构催化剂反应机理,事实上,该反应中催化剂的特殊核-壳结构没有实际贡献,核壳结构的合成是完全非必要的。
该工作为研究核壳型双金属催化剂提供了启发,例如,反应条件下核壳表面是否真实存在,是否贡献催化活性?又如,催化剂制备中追求构建核壳表面是否有必要?同时,该工作也体现了原位研究对于研究催化过程中构效关系的重要性。
中科院大连化物所张晓奔博士生、南方科技大学韩韶波博士生、中科院上海高等研究院朱倍恩副研究员、大连理工张光辉副教授为共同第一作者。中科院上海高等研究院高嶷研究员、南方科技大学谷猛副教授与中科院大连化物所刘伟副研究员、杨冰副研究员为共同通讯作者。相关成果发表在《Nature Catalysis》上,文章链接:https://www.nature.com/articles/s41929-020-0440-2。该工作得到了国家自然科学基金、大连市人才项目、中科院“从0到1”原始创新项目、中科院青年创新促进会等项目的资助,尤其得到了苏党生研究员生前的大力支持。(上海光源科学中心供稿)
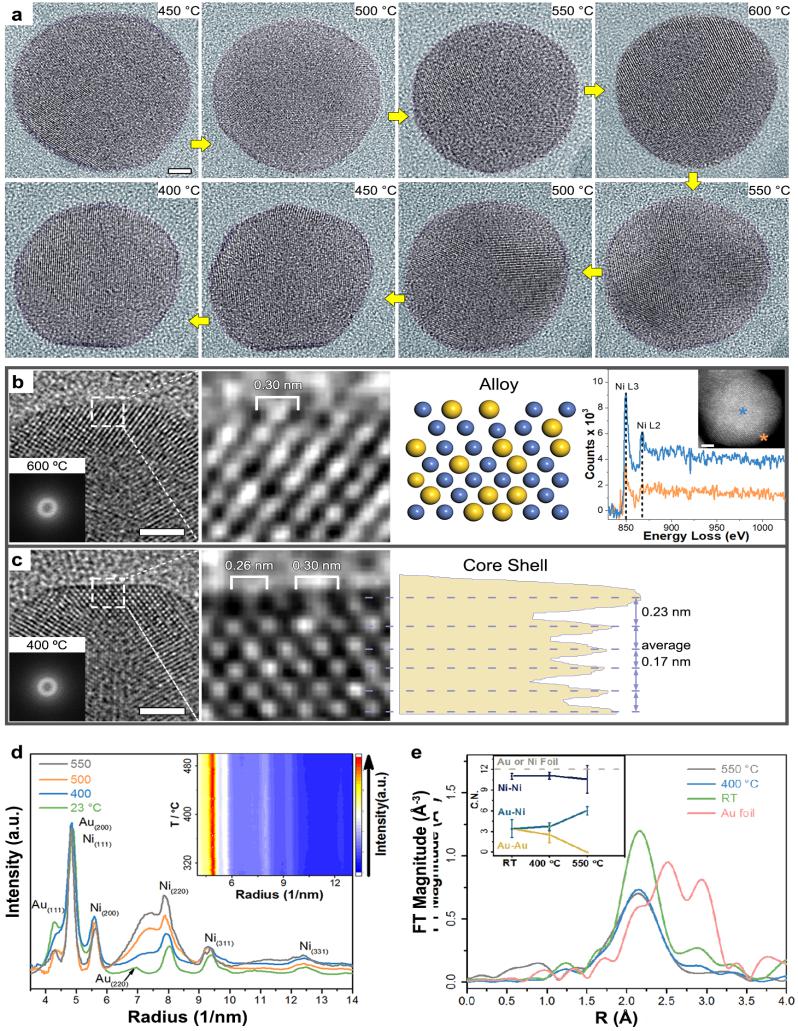
图3,原位电镜实验观察到的金镍合金核壳结构在二氧化碳加氢反应中的动态变化

图4,理论计算的不同反应物及中间产物对金镍合金表面偏析的影响、一氧化碳在金镍合金表面吸附红外谱线以及金镍合金表面反应路径计算。